Protein Determination Methods¶
Protein determination methods can be direct (test for specific features/functionality that is specific to proteins) or indirect (test for general compositional features that infer the presence of proteins)
Indirect - Kjeldahl Process - Methodology¶
Is a process that can determine the crude protein content by looking at how much nitrogen is in the sample. It is carried out in three steps
1 - Digestion¶
First the crude sample is digested with hot sulphuric acid to decompose it in to ammonium:
2 - Distillation¶
The digested protein is then deprotonated to ammonia and is absorbed into either a \(\ce{B(OH)3}\) (boronic acid) solution or a \(\ce{H2SO4}\) solution, forming ammonium again
3 -Titration¶
If the distillate was absorbed into \(\ce{B(OH)3}\), a direct titration can happen with a strong acid, showing how much of the \(\ce{B(OH)3}\) was converted into \(\ce{B(OH)4-}\) by the \(\ce{NH3 -> NH4+}\) :
If the distillate was absorbed into \(\ce{H2SO4}\) then the solution needs to be back titrated (a known amount of the absorbing solution must have been used) to determine how much the pH of the \(\ce{H2SO4}\) solution has changed.
Indirect - Kjeldahl Process - Calculation¶
Once we have the raw titrated amount of nitrogen, we can calculate how much protein that correlates to by using the Jones Factor.
We start by calculating the percentage of nitrogen in the initial sample and multiplying it by the Jones factor, which is the amount of nitrogen that you’d find in protein.
Proteins are about 16% nitrogen, though each specific food type will have a different Jones factor based on the composition of the proteins.
This has the major drawback that all nitrogen found in the sample is assumed to be from protein.
Indirect - Formol titration¶
First, the sample is treated with formaldehyde to mask the nitrogen end of the protein:
This converts the protein from a zwitterion into a purely acidic one that can be titrated with NaOH to determine the amount of the protein.
This is much faster than the Kjeldah process, but it is only useful for large quantities of proteins. the method is typically used for the determination of protein content of milk.
Colorimetric Analysis¶
These methods use the Beer-Lambert lambert law to quantitatively determine the protein content by measuring how much of a coloured product/complex is formed using spectrophotometry. As a result, standard curves are incredibly important for these methods.
They typically involve either copper chelation or protein-dye binding.
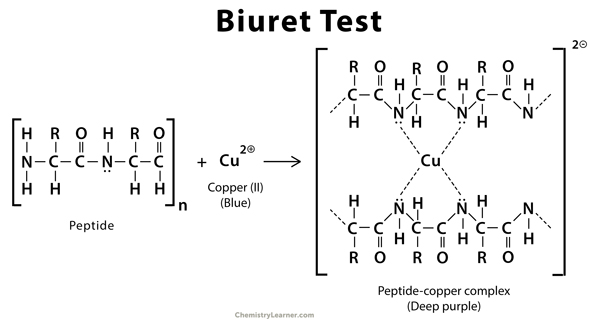
Direct - Biuret Test  ¶
¶
\(\ce{Cu(II)}\) ions form a complex with the peptide bonds in an alkaline solution that absorbs strongly at \(\lambda=540\: nm\). One copper ion forms a complex with 4-6 peptide bonds.
This particular test is sensitive between a range of \(5-160\:mg/mL\), which is useful for industrial applications, but is not sensitive enough for protein research.
Direct - Lowry Assay¶
Based on the Biuret reaction but the copper ions are produced by the oxidation of peptide bonds and the reduction of aromatic residues (tryptophan and tyrosine). this produces a dark blue molecule that absorbs strongly at \(\lambda=660\:nm\).
The reagent is sensitive within the range of \(10-1000\:\mu g/mL\) so is useful for research settings, however is sensitive to surfactants and reducing agents.

Direct - Protein-dye Binding¶
Coomassie G-250 is a dye that is used for the detection and determination of total protein content. It is typically used as a protein stain for agarose gel electrophoresis. In an acidic environment, it binds to proteins, causing it to shift colour from reddish/brown to blue at \(\lambda=575-615\:nm\).
In acidic conditions the protonated form of the dye donates an electron to free ionisable groups of the protein, denaturing it in the process, exposing hydrophobic regions and causing the dye to shift from \(\lambda=465\:nm\) to \(\lambda=595\:nm\). The hydrophobic regions of the protein then attract the hydrophobic regions of the dye. This bond is further stabilised by the positively charged (protonated) amines of the protein, lining up with the negatively charged sulphonate (\(\ce{SO3-}\)) groups of the dye.
The binding is quite fast, but on its own, but won’t bind to free amino acids or low MW proteins.
Direct - Bradford Assay¶
When Coomassie G-250 is used in an assay format it binds mostly to aromatic residues (histidine, tryptophan, tyrosine, phenylalanine) and to arginine with the same absorbance as listed above.
Hydrolysis and Ninhydrin¶
Ninhydrin is a reagent that when heated de-aminates the proteins, forming “Ruhemann’s purple” which can be spectroscopically quantified. it is extremely sensitive, however forms a yellow colour in the presence of proline,

UV-Vis Spectroscopy¶
Aromatic residues will absorb light at \(\lambda=\sim280\:nm\), meaning that all proteins that contain these residues can be quantified directly, without the need for reagents.
This process is sensitive to \(0.1-100\:\mu g/mL\) and is fast, however it is prone to contamination from other aromatic species, such as nucleic acids. As with almost all of the tests, it is incompatible with detergents and reducing agents.
